
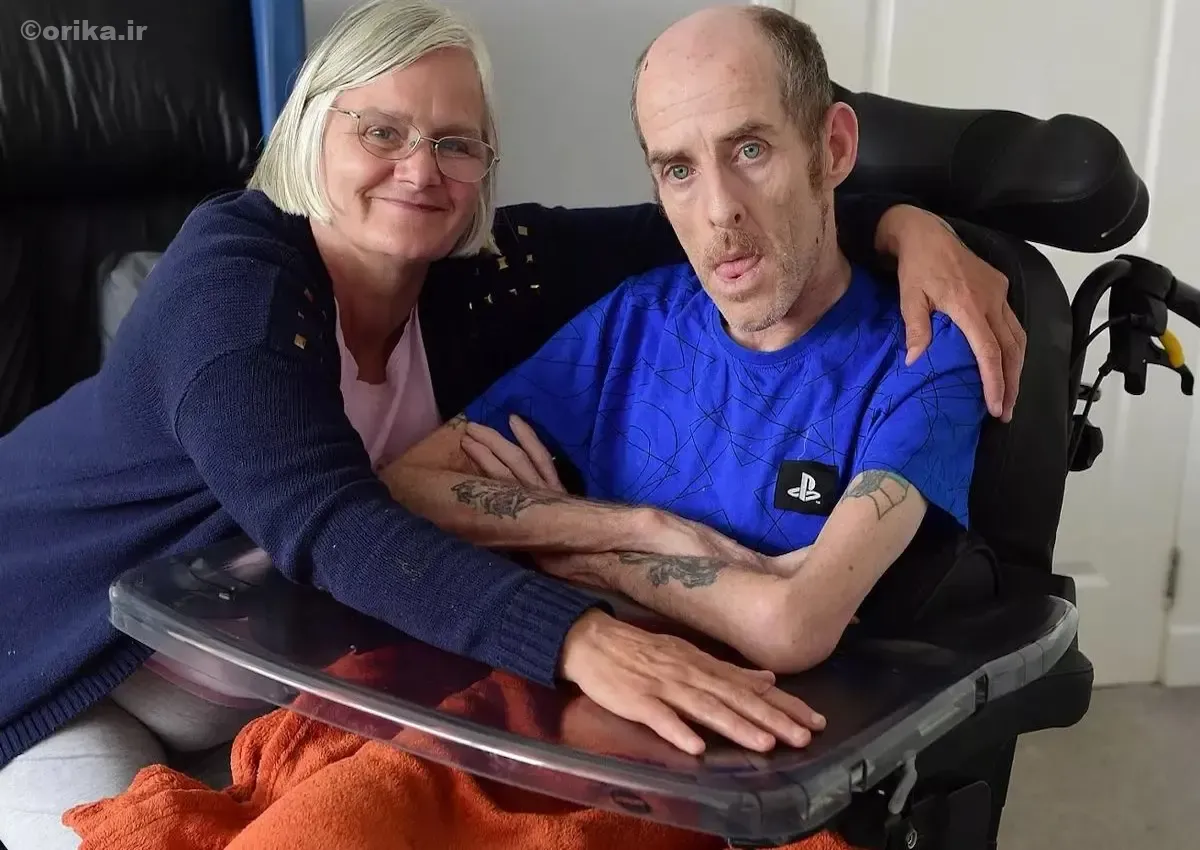
بیماری هانتینگتون (Huntington’s Disease) یک اختلال عصبی-ژنتیکی اتوزوم غالب است که بهصورت تدریجی سلولهای عصبی مغز را تخریب میکند. علت آن جهش در ژن HTT روی کروموزوم ۴ است که باعث افزایش غیرطبیعی تکرارهای CAG میشود. در نتیجه، پروتئینی سمی به نام هانتینگتین (Huntingtin) تولید میگردد. این آسیب ابتدا در استریاتوم و سپس در بخشهای شناختی و روانی مغز گسترش مییابد و سه دسته علائم ایجاد میکند: حرکتی، شناختی و روانی. بیماری معمولاً بین ۳۰ تا ۵۰ سالگی شروع میشود و میانگین امید به زندگی بیماران بین ۱۰ تا ۲۰ سال پس از بروز علائم است. نوع نوجوانی (JHD) شکل شدیدتر و سریعتر آن است که قبل از ۲۰ سالگی بروز میکند.
بیماری هانتینگتون چیست؟
هانتینگتون یکی از شناختهشدهترین بیماریهای نورودژنراتیو با منشأ ژنتیکی است. این اختلال بهصورت تدریجی عملکرد مغز را مختل میکند و در نهایت باعث از دسترفتن کنترل حرکت، گفتار، و حافظه میشود. هر فردی که نسخه معیوب ژن HTT را از یکی از والدین به ارث ببرد، احتمال ابتلا دارد. بیماری با الگوی اتوزوم غالب منتقل میشود، یعنی وجود تنها یک نسخه از ژن جهشیافته برای فعال شدن بیماری کافی است.
روند بیماری پیشرونده است و توقف ندارد اما میتوان آن را با مراقبتهای تخصصی کند کرد. علائم اولیه شامل نوسانات خلق، بیقراری و حرکات غیرارادی است که در طول سالها به ناتوانی شدید تبدیل میشوند. بسیاری از بیماران در مراحل اولیه دچار ویژگی خاصی به نام “Anosognosia” میشوند؛ یعنی توانایی درک بیماری خود را از دست میدهند. این ویژگی، درمان و همکاری با تیم مراقبی را دشوار میکند.
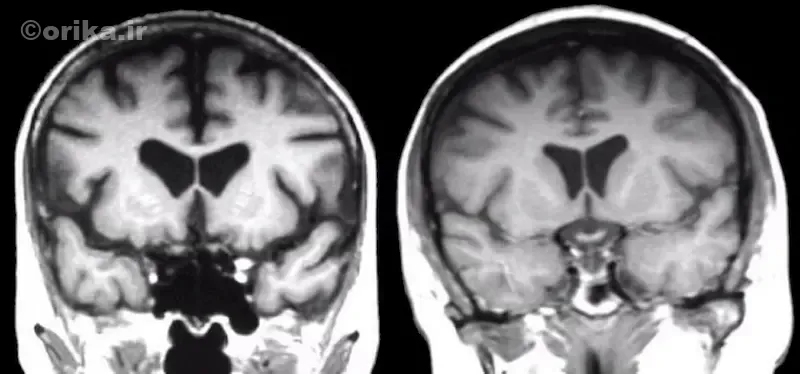

علت ژنتیکی و مولکولی بیماری
ژن معیوب HTT بهطور طبیعی در حالت سالم دارای ۱۰ تا ۲۶ تکرار CAG است. اما زمانیکه تکرارها بیشتر از حد خاصی شود، ژن توانایی تولید پروتئین سالم را از دست میدهد. تعداد تکرارهای CAG شدت و سن شروع بیماری را تعیین میکند:
| تعداد تکرار CAG | وضعیت ژن | احتمال بروز بیماری | توضیح |
|---|---|---|---|
| ≤26 | طبیعی | بدون خطر | عملکرد ژن طبیعی |
| 27-35 | بینابینی | اغلب بدون علائم | احتمال افزایش در نسل بعد |
| 36-39 | نفوذپذیری جزئی | شروع خفیف یا دیرهنگام | ممکن است بدون علامت باقی بماند |
| ≥40 | نفوذپذیری کامل | ابتلا قطعی | شروع بیماری در جوانی یا میانسالی |
پدیدهای به نام Anticipation سبب میشود که در نسلهای بعد، تکرارهای CAG افزایش یابد و بیماری زودتر و شدیدتر شروع شود. این پدیده بیشتر هنگام انتقال از پدر به فرزند رخ میدهد. تجمع پروتئینهای سمی هانتینگتین ارتباط سلولهای عصبی را مختل کرده و در نهایت منجر به مرگ آنها میشود.
علائم بیماری هانتینگتون
هانتینگتون سه دسته علائم اصلی دارد که بهمرور شدت میگیرند:
- علائم حرکتی: شامل حرکات بیاراده (کره یا Chorea)، دیستونی، مشکلات تعادل، لرزش، و دشواری در بلع است.
- علائم شناختی: کاهش تمرکز، ضعف تصمیمگیری، کندی تفکر و اختلال حافظه. این موارد موجب افت عملکرد شغلی و اجتماعی میشوند.
- علائم روانی: افسردگی، اضطراب، تحریکپذیری، بیانگیزگی، وسواس رفتاری و گاهی رفتارهای پرخشم یا پرخطر.
بیماران معمولا در مراحل پیشرفته قدرت گفتار و تعادل را از دست میدهند. در نوع نوجوانی (JHD) کره حرکتی کاهش یافته و سفتی عضلات، تشنج و تحلیل سریع ذهنی دیده میشود. برخی بیماران دچار بیخوابی مزمن و کاهش وزن غیرقابل توضیح میشوند، حتی با تغذیه کافی. این پدیده بهدلیل هایپرمتابولیسم مغزی است که یکی از نکات کمتر شناختهشده بیماری محسوب میشود.
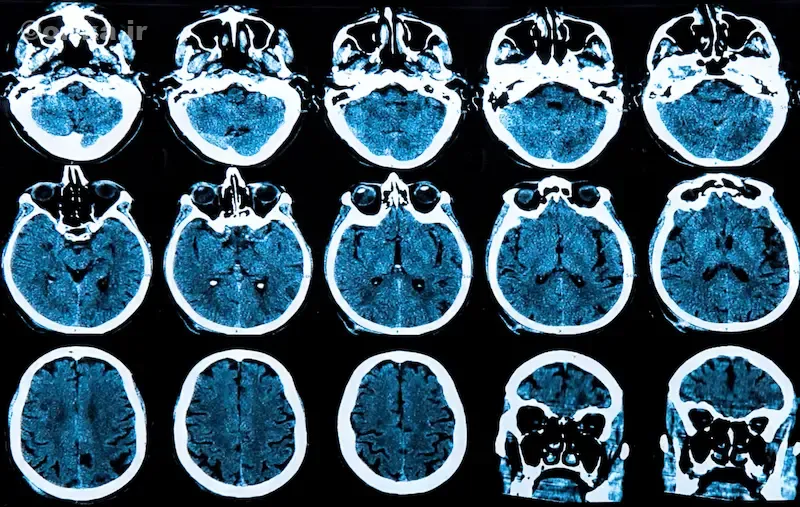

پاتوفیزیولوژی و آسیبشناسی
تخریب عصبی در مراحل اولیه از استریاتوم آغاز میشود؛ ناحیهای که در کنترل حرکت و احساس پاداش نقش دارد. در پی تحلیل تدریجی سلولهای این بخش، حرکات بدن از کنترل خارج میشوند. MRI نشاندهنده آتروفی هسته دمدار (Caudate nucleus) و بزرگشدن بطنهای جانبی است.
اختلال در انتقالدهندههای عصبی GABA و گلوتامات سبب کاهش مهار در مدارهای حرکتی و بروز حرکات غیرارادی میشود. همچنین، کاهش تولید فاکتور رشد نورونی BDNF موجب تضعیف ارتباط قشر فرونتال و عقدههای قاعدهای میشود. این فرآیند برگشتپذیر نیست و بهمرور موجب تحلیل ذهن و تغییر رفتار میگردد.
در مراحل پایانی، مغز بهطور قابل توجهی کوچک میشود و وزن آن تا ۳۰٪ کاهش مییابد. همبستگی مستقیم بین شدت آتروفی و تعداد تکرارهای CAG ثابت شده است.
تشخیص بیماری هانتینگتون
تشخیص با بررسی ترکیب علائم بالینی، سابقه خانوادگی و آزمایش ژنتیکی انجام میگیرد. تست DNA تعداد تکرارهای CAG را مشخص میکند و نتیجه قطعی ارائه میدهد. سیستم امتیازدهی UHDRS (Unified Huntington’s Disease Rating Scale) شدت بیماری را از نظر حرکات، شناخت و رفتار ارزیابی میکند.
پزشک متخصص مغز و اعصاب معمولاً از MRI و گاهی PET-scan برای بررسی آتروفی مغز استفاده میکند. در مراحل اولیه، تغییرات تصویری شامل باریکشدن استریاتوم و افزایش فضاهای بطنی است. مهم است که بیماریهای مشابه مانند HDL2، SCA17، Wilson و parkinsonism از هانتینگتون افتراق داده شوند. قطعیت تشخیص، فقط با تست ژنتیک مستقیم HTT CAG ممکن است.

راه های درمان و مدیریت بیماری
درمان قطعی وجود ندارد اما رویکرد چندرشتهای (Multidisciplinary) میتواند علائم را کنترل کند. درمان شامل دارو، توانبخشی و حمایت روانی است.
- داروهای حرکتی: Tetrabenazine یا Deutetrabenazine حرکات کره را کنترل میکنند (مهارکننده VMAT2).
- داروهای روانی: SSRIs برای افسردگی و Olanzapine/Risperidone برای تحریکپذیری یا روانپریشی.
- توانبخشی: فیزیوتراپی جهت تقویت تعادل، گفتاردرمانی برای جلوگیری از خفگی و کاردرمانی برای حفظ استقلال در زندگی روزمره.
- تغذیه و سبک زندگی: رژیم پرکالری ضروری است زیرا بیماران دچار سوختوساز بالا هستند. همچنین، محیط امن و حمایت مداوم برای کاهش خطر Self-harm حیاتی است.
با رعایت این نکات، پیشرفت علائم کندتر میشود و کیفیت زندگی تا بیش از یک دهه حفظ میگردد.
وراثت و مشاوره ژنتیک
هانتینگتون با الگوی اتوزوم غالب منتقل میشود؛ هر فرزند فرد مبتلا ۵۰٪ شانس ابتلا دارد. مشاوره ژنتیک برای خانوادههای در معرض خطر ضروری است. آزمایش پیش از بارداری با PGT-M یا آمنیوسنتز میتواند از تولد فرزند مبتلا جلوگیری کند.
در برخی موارد، افراد تصمیم میگیرند نتیجه آزمایش خود را ندانند (non-disclosing testing) تا از اضطراب روانی جلوگیری شود. مرحله مشاوره باید شامل توضیح کامل ریسک، پدیده Anticipation، و توصیههای مراقبتی باشد. آگاهی از سابقه خانوادگی، نخستین گام در تشخیص زودهنگام بیماری هانتینگتون است.
چشمانداز و تحقیقات جدید درباره هانتینگتون
امید به زندگی بیماران بین ۱۰ تا ۲۰ سال پس از شروع علائم است. علت مرگ بیشتر پنومونی، سوءتغذیه یا عفونتهای مزمن است. تحقیقات نوین روی تکنیکهای RNA interference (RNAi) و antisense oligonucleotides (ASOs) متمرکز شدهاند تا بیان ژن HTT را خاموش کنند.
در کنار آن، مطالعات روی افزایش BDNF و کنترل استرس اکسیداتیو با ترکیبات Resveratrol و CoQ10 در دست بررسی است. گرچه هیچ درمانی تاکنون روند بیماری را متوقف نکرده، اما امید به بهبود در سطح سلولی در آینده نزدیک وجود دارد. مسیر تحقیق جهانی بر درمان هدفمند و شخصیسازیشده مبتنی بر پروفایل ژنتیکی بیماران پیش میرود.
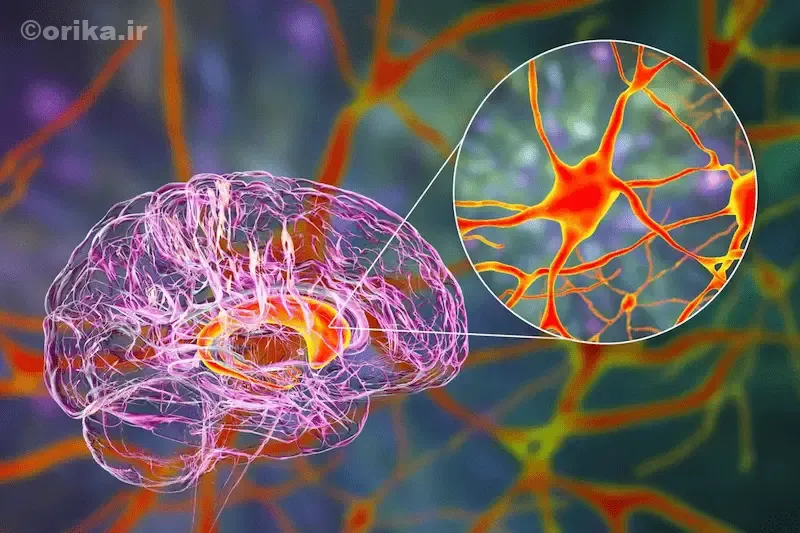
نتیجهگیری
هانتینگتون یک بیماری ژنتیکی پیچیده است که پایه آن بر تخریب سلولهای عصبی قرار دارد. تشخیص زودهنگام، مشاوره ژنتیک، و مراقبت چندرشتهای میتوانند پیشرفت بیماری را کند کنند. شناخت دقیق ژن HTT و تعداد تکرارهای CAG اهمیت حیاتی دارد. توجه به سلامت روانی بیمار، همراهی خانواده و تغذیه مناسب مؤثرترین راههای مدیریت بیماریاند. امید علمی به درمانهای ASO و RNAi نشانگر آیندهای روشنتر برای مبتلایان است.
در میان اختلالات روانی مرتبط با بیماری هانتینگتون، گاهی نشانههایی بروز میکند که مرز واقعیت و خیال را در ذهن فرد مبهم میسازد. این وضعیت، ریشهای ژنتیکی و عصبی دارد و درک عمیق آن کلید درمان مؤثر است. در مجله اوریکا تحلیلی علمی و روانشناختی درباره اینکه روانپریشی (سایکوتیک) چیست؟ منتشر شده است؛ مطالعه این بخش دید تازهای به پیوند پیچیده میان تخریب سلولهای عصبی و بروز رفتارهای روانپریشانه میدهد—روایتی دقیق، متفاوت و ضروری برای هر خواننده علاقهمند به سلامت روان و علوم عصبی.
سوالات متداول
- سن شروع بیماری هانتینگتون چقدر است؟ معمولاً بین ۳۰ تا ۵۰ سالگی، ولی نوع نوجوانان قبل از ۲۰ سال بروز میکند.
- آیا بیماری ارثی است؟ بله، با الگوی اتوزوم غالب از یکی از والدین منتقل میشود.
- آیا تست ژنتیکی قطعی است؟ بله، آزمایش تعداد تکرارهای CAG نتیجه قطعی را نشان میدهد.
- امید به زندگی بیماران چقدر است؟ بین ۱۰ تا ۲۰ سال پس از شروع علائم.
- آیا درمان قطعی وجود دارد؟ فعلاً نه، ولی درمانهای علامتی و ژنتیکی جدید در حال توسعهاند.
-

-

-

-

-

-

-

-

-

-

-

-
-
-

-
-

-

-

-
-
-

-
-
-
-

-
-
-
-

-

-
-

-
-
-
-

-

-

-
-
-
-
-
-

-

- هورمون پرولاکتین در مردان؛ علائم، علل و درمان کامل
-

-

-

-
-

-
-

-

-
-

-

-
-
-

-

-

-
-

-

-

-

-

-

-

